| Issue |
J Oral Med Oral Surg
Volume 27, Number 1, 2021
|
|
|---|---|---|
| Article Number | 16 | |
| Number of page(s) | 8 | |
| DOI | https://doi.org/10.1051/mbcb/2020048 | |
| Published online | 11 December 2020 | |
Educational Article
Dermatomyositis: what the oral healthcare provider must know
1
Department of Oral and Maxillofacial Surgery, School of Dental Sciences, Sharda University, Plot 32, 34 Knowledge Park 3,
Greater Noida 201308, Uttar Pradesh, India
2
COSMOZONE Dental and Implant Clinic, Greater Noida, Uttar Pradesh, India
* Correspondence: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
14
May
2020
Accepted:
26
August
2020
Abstract
Dermatomyositis (DM) is an autoimmune condition characterized by skin rashes and progressive muscle weakness. It is classified under the idiopathic inflammatory myopathies (IIM) and can affect children as well as adults. A heightened incidence of malignancy in adults with DM has laid greater focus on its early diagnosis, treatment, and monitoring. In recent years, a greater understanding of the pathogenesis of the disease, its diagnostic criteria and management has improved the quality of life in affected individuals. The orofacial region presents with many manifestations of the disorder, sometimes even the initial presenting signs. This review presents an update on the disease process, its pathogenesis, diagnostic criteria, orofacial manifestations, medical management and dental considerations for patients with DM. The updated knowledge about DM is crucial for oral health care providers to plan and execute oral health care in a coordinated manner.
Key words: Dermatomyositis / oral manifestations / facial rash / management / dental considerations / malignancy
© The authors, 2020
 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
Idiopathic inflammatory myopathies (IIMs) are a group of heterogeneous disorders affecting the skeletal muscles, often with coexistent extramuscular manifestations such as interstitial lung disease (ILD), arthritis and malignancies [1]. Dermatomyositis (DM) is the most common subset of IIMs, the others being Polymyositis (PM) and inclusion body myositis (IBM). Necrotizing autoimmune myositis (NAM) has been recently included as a distinct myositis subset [2]. DM is an autoimmune disorder characterized by progressive muscle weakness, with recent emphasis being laid on its significant morbidity and mortality [3]. Orofacial manifestations are significant findings in DM and so are of great relevance to oral health care providers.
DM was first reported in 1875 by Poten [4] while the term ‘dermatomyositis’ was coined by Unvericht in 1891 [5]. Diagnostic criteria and classification of DM have continually evolved in an effort to distinguish DM from similar autoimmune disorders, especially when muscle weakness is weak or absent and when skin lesions resemble those observed in other autoimmune connective tissue diseases such as systemic lupus erythematosus (SLE). Adult DM patients have exhibited an increased risk of development of malignancy, an observation that has made early diagnosis, treatment and continual monitoring crucial [6].
This review focuses on the latest developments in the diagnosis of this disease, its orofacial manifestations, medical management and dental considerations for patients with DM. Updated knowledge about DM is crucial for oral health care providers to plan and execute oral health care in a coordinated manner.
Epidemiology
The rarity of the condition has lead to limited information on its incidence and prevalence. The reported incidence in literature is 9.63 cases per million persons, while clinically amyopathic DM (ADM) is estimated to be 2.08 cases per million. DM exhibits a bimodal age distribution of children under 18 years (juvenile DM, JDM) and adults in their late 40s to early 60s [7]. There is a female predominance over males with a ratio of 1.5–2.0:1; however, in older patients with malignancy, there is an equal sex ratio. Diagnosis is generally made at an average age of 40 years, while malignancy is commonly observed at an average age of 55 years. DM associated with a connective tissue disease is predominantly observed among younger women with a higher prevalence in African-Americans [7].
Etiopathophysiology
The current belief in describing the pathophysiology of DM is that the disorder develops in genetically susceptible individuals as a result of an autoimmune attack on specific organs, most likely being triggered by environmental factors [8]. The endothelium of endomysial blood vessels is thought to be the site of early pathogenic events. Once the activation of C3 (complement 3) by antibodies or other factors is complete, C3b and C4b fragments are formed. This event is followed by the formation of C3bNEO (a neoantigen expressed on the surface of activated C3 component) and membrane attack complex (C5b-C9), which are deposited in the endomysial vasculature [8]. Perivascular inflammation, loss of capillaries, muscle ischemia and perifascicular muscle fibre atrophy occur as a result of this deposit, which in turn results in muscle atrophy [6]. Pathophysiology of skin lesions, although not fully understood, is believed to follow similar mechanisms. Genetic associations with HLA DRB1*0301 and DQA1*0501 in Caucasians and HLA-B7 in Asians have fuelled genetic predisposition theories. [8] Proof of this association is even greater in DRB1*0301 or DQA1*0501 haplotypes with the autoantibody anti-Jo1 and DRB1*07 or DQA*0201 haplotypes with anti-Mi-2 antibodies [8]. HLA-B8, HLA-DR3 and HLA-DQA1*0501 are the most common allelic associations found in juvenile DM (JDM), while HLA-B18 and HLA-B35 are common in drug-induced DM [8].
An alternate mechanism for perifascicular myofibre injury was proposed by Greenberg et al. in 2005, where they implicated chronic overproduction of interferon alpha/beta inducible proteins [9]. This theory was supported by the findings of Kim et al. [10]. Tumor necrosis factor alpha (TNF-α) has also been implicated in the pathogenesis of DM [11]. Environmental factors, such as changing season, ultraviolet light exposure, drugs (hydroxyurea, penicillamine, statin drugs, quinidine and phenylbutazone), infectious agents such as Toxoplasma, Borrelia, coxsackie virus, parvovirus, echovirus, human T-cell lymphotropic virus (HTLV-1), Human immunodeficiency virus (HIV), Hepatitis B and C viruses, filler materials (silicone) [12] and certain lifestyle factors have been suggested as triggering factors [13].
Clinical features
Cutaneous manifestations are a feature in 30–40% of adult patients with classical DM while in JDM cases the percentage involvement is 95% [8]. These are highly suggestive of the diagnosis of DM, owing to their characteristic clinical features. In 60% of cases, cutaneous signs precede muscle involvement, while they may appear simultaneously with or follow development of myositis. Amyopathic DM (ADM) refers to a small percentage (4–8.2%) of patients with DM who never develop myositis, but exhibit exclusive skin disease. The cutaneous manifestations of DM are enumerated in Table I [14].
Systemic features associated with DM include proximal muscle weakness that affects the upper arms, neck and legs, usually in a symmetric pattern [6]. Symptoms may range in severity from difficulty in raising arms, leading to impairment of daily gestures to difficulty climbing stairs or at worst, even getting up from bed or from a chair to standing position (Gower's Sign). In progressive stages of the disease, orthotic or other assistive devices may be required to cater to the physical disabilities such as the Trendelenberg sign (diagnostic test of hip dysfunction due to abductor muscle weakness). Respiratory failure may develop in severe cases of DM.
Interstitial lung disease (ILD), leading to severe dyspnoea and possible progression to pulmonary insufficiency, is present in 10–45% of patients with classical and amyopathic DM [8]. Polyarthralgia or symmetric polyarthritis, mostly affecting the hands, wrists, shoulders, ankles, and knees is typically observed in patients exhibiting symptoms that overlap with connective tissue disorders. Cardiac involvement is seen in 1/3rd of DM cases, mostly subclinical in nature, though clinical disease may present as congestive heart failure.
DM is thought to be a paraneoplastic phenomenon, with malignancy preceding or following the development of clinical signs of DM in 2.5–29% cases [15]. Solid organ malignancy is most commonly associated with DM, a majority of cases involving the breast, ovary, uterus, cervix, colon, rectum, lung and prostate. Nasopharyngeal carcinoma is commonly observed among Southeast Asians. The highest incidence of malignancy is found in the first year after the development of DM symptoms. However, the first three years after symptom onset are associated with an elevated risk of malignancy, particularly in elderly males [8]. Fortunately, there does not appear to be an association between JDM and malignancy.
Facial and oropharyngeal manifestations of JDM and DM are listed in Table II [7,16–20]. The scalp may present with non-scarring alopecia, especially following a disease flare up. Diffuse, scaly, psoriasis-like lesions, but with poikilodermatous characteristics and intense pruritis are observed. These features present a diagnostic dilemma with seborrheic dermatitis, lupus erythematosus or psoriasis, eventually requiring histologic evaluation for diagnosis [19]. Scalp dysesthesia may be observed even without any eruptions [20]. Midfacial erythema involving the nasolabial folds is a feature that can be distinguished from the malar erythema (sparing the nasolabial folds) typically associated with acute cutaneous lupus erythematosus [20].
Cutaneous manifestations of dermatomyositis.
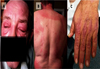 |
Fig 1 (A) Heliotrope rash of the eyelids. (B) The shawl sign over the upper back. (C) Gottron's papules over the extensor surfaces of the metacarpophalangeal and interphalangeal joints, with involvement of adjoining skin. (Reprinted from Chu LL, Rohekar G. Dermatomyositis. CMAJ. 2019; 191(12):E340. doi:10.1503/cmaj.180947, with permission). |
Orofacial manifestations of dermatomyositis.
Diagnosis
Diagnostic criteria and classification of DM have been continuously revised since the first criteria were proposed by Bohan and Peter in 1975, which was based on their clinical experience rather than on specific objective criteria [6]. Most recently, the European League Against Rheumatism/American College of Rheumatology (EULAR/ACR) classification criteria for idiopathic inflammatory myopathies (IIMs) was validated in 2017 [21]. Accordingly, conditions included as a subset of IIMs are juvenile dermatomyositis (JDM), dermatomyositis (DM), amyopathic dermatomyositis (ADM), hypomyopathic dermatomyositis (HDM), inclusion body myositis (IBM), polymyositis (PM) and immune-mediated necrotizing myopathy (IMNM) and juvenile myositis other than JDM.
Diagnosis of DM revolves around assessment of classic skin manifestations, proximal muscle weakness, laboratory investigations such as elevated serum creatine kinase (CK) levels (indicative of muscle damage) and muscle biopsies, magnetic resonance imaging (MRI), ultrasonography (USG) and electromyography (EMG) [21]. Muscle biopsy typically reveals endomysial infiltration of mononuclear cells surrounding, but not invading, myofibers, perifascicular atrophy and rimmed vacuoles [21]. Five myositis-specific autoantibodies (MSA) have been identified, namely anti–Jo-1, anti– Mi-2, anti–signal recognition particle, anti–PL-7, and anti–PL-12, all of which are strongly associated with particular subgroups of IIMs [22]. Among these, anti-Mi-2 antibodies are considered DM specific and show good response to therapy [6].
Medical management
DM is incurable, therefore, management of symptoms, improvement of functions and prevention of disability form the therapeutic objectives. A multidisciplinary team approach involving rheumatologists, dermatologists and pulmonologists along with patient reassurance and education, as well as periodic assessment of symptoms are particularly important.
The Japan College of Rheumatology, Japanese Society of Neurology and Japanese Dermatological Association established a consensus on the treatment of DM and JDM in 2018 [3]. Levels of evidence and recommendation grades used in this consensus are listed in Table III [3]. Glucocorticoids (GC) is the preferred first line of therapy in DM (evidence level: VI, recommendation grade: B) [23]. Prednisolone is used in doses of 0.75–1 mg/kg per day orally for 3–4 weeks, followed by tapering dose to once daily or alternate-day schedule, with the goal of stopping completely in 2 years (recommendation grade: C1). Complete remission rate after the termination of medication has been reported to vary from 25% to 87% [24]. Side effects of GC therapy include steroid myopathy (evidence level: IV) [3], moon face, diabetes, central obesity, gastric irritation, psychiatric changes, dermatological symptoms and osteoporosis (evidence level III) [3]. In JDM, initial therapy with combination GC and methotrexate (MTX) 15–20 mg/m2 is used with a goal of early reduction of corticosteroids (evidence level V) [25]. Side effects of MTX therapy are liver damage, lung fibrosis, leukopenia, infection, neoplasia, stomatitis and gastric irritation [6].
Some patients do not respond to GC alone, while other patients treated with GC alone experience recurrence after tapering of GC doses. Immunosuppressants are chosen in patients with DM who are resistant to GC therapy (recommendation grade: B) [3]. Choice of immunosuppressant can be among MTX (7.5–15 mg/week), azathioprine (AZA, 50–100 mg/day) [6], tacrolimus (Tac, trough concentration 5–10 ng/ml or 0.075 mg/kg/day) [26], cyclosporin A (CsA, 4–6 mg/kg/day) [27], mycophenolate mofetil (MMF, 1–3 g/day) [3] and cyclophosphamide (CPA, 50–100 mg/day) [28]. Combination of GC and immunosuppressants is chosen with the objective of keeping high dose GC therapy limited to a short period (recommendation grade: C1) [3] and reduce relapse rates during GC dose tapering (evidence level VI) [3].
Intravenous immunoglobulin (IVIg, 2 g/kg/month) [29] can be considered as first line therapy in steroid resistant DM (recommendation grade: B) [3], especially utilized as therapy for dysphagia (evidence level: V, recommendation grade: C1) [3,30]. Biological agents are used in cases of relapse of DM, such as Tocilizumab [31] (interleukin 6 receptor antagonist, recommendation grade: C1) [3], abatacept (fully human fusion of cytotoxic T lymphocyte antigen-4 and the Fc portion of immunoglobulin, recommendation grade: C1) [3,32] and rituximab (B-cell depleting agent, no recommendation grade) [21,33]. Plasmapheresis has been used in DM therapy (recommendation grade: C2) with unconvincing results [3].
For DM patients with cutaneous manifestation alone, observation or topical corticosteroid therapy should be used [3].
Adjunctive therapies
Muscle strength and range of motion can be regained with physiotherapy [33], exercise [34] and thermal applications. Speech therapy is essential to assist patients of DM with difficulty in speech [35]. Avoidance of exposure to excessive sunlight, use of a broad-spectrum sunscreen lotion and protective clothing form part of the advice to patients to counter photosensitivity and in those with erythematous lesions in sun-exposed areas of the body. When calcinosis leads to nerve compression pain and recurrent infections, surgical removal (evidence level: V) [3], carbon dioxide laser or extracorporeal can be considered [36].
Prognosis
The use of corticosteroids and other immunosuppressants has significantly improved the prognosis of DM. Survival has improved to 79–82% at 1 year, 70–77% at 5 years and 55–66% at 10 years [37]. Improvement depends on whether aggressive therapy was initiated early in the disease process. Therefore, remission period can last as long as 20 years or more, provided initial therapy was aggressive and was tapered over two to three years while concomitantly limiting muscle disease activity and avoiding flare ups. Poor prognosis is generally associated with higher age at diagnosis, male gender, non-Caucasian race, malignancy, ILD, cardiac involvement, diabetes, thrombocytopenia and arthritis [37]. The most common causes of death are respiratory failure, in association with dysphagia related aspiration pneumonia and malignancy in adult DM. Prognosis of JBM is significantly better, with survival rates of 92% after therapy.
Several biomarkers are now utilized for clinical association and follow-up of the treatments' response, such as anti-MDA5 (Melanoma differentiation-associated gene 5), anti-NXP-2 (Nuclear matrix protein 2), type I IFN (interferon), IL-18 (interleukin), IL-6, BAFF (B cell-activating factor), ferritin, SPA/SPD (surfactant protein D), and KL6 (Krebs von den Lungen-6) [38,39].
Oral healthcare considerations
The characteristic facial and/or scalp skin rashes, as well certain oropharyngeal features may be the first manifestations of DM [37] and JDM [40]. This makes the role of the oral healthcare provider crucial in providing initial evidence of a greater underlying disease. A group of DM patients present with salivary hypofunction or xerostomia, manifest by a higher dental caries index, oral mucosal dryness and greater dental plaque accumulation [7]. Those with xerostomia may be assisted with salivary substitutes in the form of gels, mouthwashes, lozenges, oils, sprays, toothpastes and chewing gums to protect and hydrate the oral mucosa and reduce plaque accumulation and dental caries [41]. However, developments in hydrogel and buccal mucoadhesive polymer technology [42] allows for continuous release of salivary substitutes with inherent benefits for oral hydration and dental care. Another strategy for retaining artificial saliva in the oral cavity is fabrication of modified prosthetic structure in dental prosthesis in patients who wear them [43]. Salivary hypofunction requires the use of salivary stimulants such as systemic pilocarpine or cevimeline (USFDA approved) or topical sugar free chewing gums or jellybeans (mechanical salivary stimulation) [41]. Topical agents possess the added advantage of incorporating other agents such as chlorhexidine, xylitol, calcium phosphate and fluoride which reduce bacterial counts, plaque accumulation and dental caries.
Oral lichen planus (OLP) lesions that are symptomatic require topical steroid applications, although Geist et al. have reported a case of resolution of OLP following intravenous immunoglobulin therapy [44]. Oral cancers associated with DM have been reported at sites such as the tongue and tonsillar pillars. Such suspected lesions must promptly be biopsied and assessed histopathologically to establish diagnosis followed by appropriate surgical resection and/or radiotherapy. Dysphagia associated with DM must be carefully assessed and diagnosed as it affects the overall outcome of DM patients. It is associated with poor prognosis as it directly correlates to severity of muscle and pulmonary involvement, thereby increasing the risk of aspiration pneumonia (Tab. IV) [18,45]. A history of food getting stuck in the throat, difficulty in swallowing solid food, coughing during meals or having to swallow many times to clear food from the mouth are indicative of dysphagia. Such patients must not be placed in reclined position during dental treatment as this increases their chances of aspiration pneumonia [6]. For the same reason, all restorative and endodontic procedures, and single tooth extractions must be performed with rubber dam application. However, in those who exhibit severe form of dysphagia, ultrasonic oral prophylaxis, surgical removal of impacted teeth or tooth preparation for prosthetic crown fabrication may best be performed under general anaesthesia. The importance of maintenance of oral hygiene in such patients is further reinforced by the fact that dysphagia experts believe that poor oral care can lead to aspiration of oral bacteria resulting in aspiration pneumonia [46]. Oral hygiene measures including must be reinforced in such patients. Use of an electronic toothbrush could be suggested in DM patients in whom advanced muscle weakness in the extremities and fatigability make oral hygiene procedures tedious.
Dental appointments must be of short duration considering that muscle weakness involving masticatory or neck muscles could fatigue the patient on prolonged mouth opening [6]. Use of a rubber bite block may be beneficial in such patients. Surgical procedures face the risk of postoperative wound infection. It is imperative for the oral healthcare provider to plan any surgical procedure only after careful discussion about the patient's immune status with the treating physician [6]. Gastric irritation is common in patients who are on prolonged therapy with GC, MTX and MMF [6]. Therefore, the choice of medications to be prescribed must take this fact into consideration.
About 40% of DM patients manifest the ovoid palatal patch which is highly associated with the presence of anti-TIF1-γ antibodies [18]. Consequently, presence of this patch may act as a clinical identifier of patients with possible malignancy in anti-TIF1-γ antibody positive DM patients. Subclinical dental focus of infection has been reported as a prime cause/trigger of DM, which resolved after successful resolution of the infective focus and concomitant ozone therapy [47]. This demonstrates the importance of evaluation of oral health and its maintenance for a successful outcome in DM management. Bilaterally symmetrical, calcified subcutaneous nodules at the mandibular angle region have been reported in a patient with JDM [48]. Therefore, the authors suggested that any bilateral, symmetric, calcified focal points in the facial bones must be carefully investigated for the presence of a systemic disease such as DM.
Nodular dystrophic calcified aggregates may be noted within the periodontal ligament (PDL) of teeth [49]. These calcified masses would appear to be distinct from calculus deposits. They may be a novel sign of DM that can be used by oral health care providers to screen patients for definitive DM diagnostic testing and follow-up, especially in light of the fact that malignant disease is commonly associated with DM.
Subgroups of dm of interest to the oral and maxillfacial surgeon.
Conclusion
Oral healthcare providers rarely encounter DM patients, owing to the fact that it is a rare disease. However, its association with malignancy, dysphagia and orofacial manifestations has to be given due credit. It is important to recognize and understand this complex disease as oral health care providers may be the first to identify initial presenting signs of the disease. Once diagnosed, it is vital to coordinate with the patient's treating doctor for successful dental management and improvement of quality of life. As understanding of the disease process and its management principles in an ongoing process, oral health care providers must keep abreast with the latest information about DM.
Conflicts of interest
All the authors of this article report no conflict of interest regarding this work.
References
- Sasaki H, Kohsaka H. Current diagnosis and treatment of polymyositis and dermatomyositis. Mod Rheumatol 2018;28:913–921. [CrossRef] [PubMed] [Google Scholar]
- Ghirardello A, Bassi N, Palma L, Borella E, Domeneghetti M, Punzi L, et al. Autoantibodies in polymyositis and dermatomyositis. Curr Rheumatol Rep 2013;15:335. [CrossRef] [PubMed] [Google Scholar]
- Kohsaka H, Mimori T, Kanda T, Shimizu J, Sunada Y, Fujimoto M, et al. Treatment consensus for management of polymyositis and dermatomyositis among rheumatologists, neurologists and dermatologists. Mod Rheumatol 2019;29:1–9. [CrossRef] [PubMed] [Google Scholar]
- Poten PC. Morve chronique de forme anormal. Bull et Mem Hop Paris. 1875;12:314–318. [Google Scholar]
- Unverricht H. Dermatomyositis acuta. Dtsch Med Wochenschr 1891;17:41–44. [CrossRef] [Google Scholar]
- Tanaka TI, Geist SM. Dermatomyositis: a contemporary review for oral health care providers. Oral Surg Oral Med Oral Pathol Oral Radiol 2012;114:e1–8. [CrossRef] [PubMed] [Google Scholar]
- Heath KR, Fazel N. Oral Signs of Connective Tissue Disease. In Oral Signs of Systemic Disease 2019 (pp. 91–112). Cham: Springer. [CrossRef] [Google Scholar]
- Bogdanov I, Kazandjieva J, Darlenski R, Tsankov N. Dermatomyositis: current concepts. Clin Dermatol 2018;36:450–458. [CrossRef] [PubMed] [Google Scholar]
- Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol 2005;57:664–678. [CrossRef] [PubMed] [Google Scholar]
- Kim JS, Bashir MM, Werth VP. Gottron's papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: a unique molecular signature. J Invest Dermatol 2012;132:1825–1832. [CrossRef] [PubMed] [Google Scholar]
- Nabatian AS, Bashir MM, Wysocka M, Sharma M, Werth VP. Tumor necrosis factor alpha release in peripheral blood mononuclear cells of cutaneous lupus and dermatomyositis patients. Arthritis Res Ther 2012;14:R1. [CrossRef] [PubMed] [Google Scholar]
- Hu HC, Cho HY, Chiu YH. Dermatomyositis induced by filler rhinoplasty using liquid silicone. JAMA Otolaryngol 2020;146:205–206. [Google Scholar]
- Maan MA, Akhtar SJ, Haque H. Dermatomyositis. J Pakistan Assoc Dermatol 2016;18:33–43. [Google Scholar]
- Mainetti C, Terziroli Beretta-Piccoli B, Selmi C. Cutaneous manifestations of dermatomyositis: a comprehensive review. Clin Rev Allergy Immunol 2017;53:337–356. [CrossRef] [PubMed] [Google Scholar]
- Adi AH, Alturkmani H, Spock T, Williams Yohannes P, Wargo S, Szabo E, et al. Dermatomyositis paraneoplastic syndrome before symptomatic tonsillar squamous cell carcinoma: a case report. Head Neck 2015;37:E1–E3. [CrossRef] [PubMed] [Google Scholar]
- Ruparelia P, Verma O, Shah V, Shah K. Juvenile Dermatomyositis − a case report with review on oral manifestations and oral health considerations. Int J Orofacial Myol 2018;44:52–61. [Google Scholar]
- Dourado MR, da Silva Filho TJ, Salo T. Oral signs in juvenile dermatomyositis. J Oral Diag 2017;2:1–5. [CrossRef] [Google Scholar]
- Bernet LL, Lewis MA, Rieger KE, Casciola-Rosen L, Fiorentino DF. Ovoid palatal patch in dermatomyositis: a novel finding associated with anti-TIF1γ (p155) antibodies. JAMA Dermatol. 2016;152:1049–1051. [CrossRef] [PubMed] [Google Scholar]
- Callen JP. Cutaneous manifestations of dermatomyositis and their management. Curr Rheumatol Rep 2010;12:192–197. [CrossRef] [PubMed] [Google Scholar]
- Cobos GA, Femia A, Vleugels RA. Dermatomyositis: an update on diagnosis and treatment. Am J Clin Dermatol 2020;21:339–353. [CrossRef] [PubMed] [Google Scholar]
- Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, de Visser M, et al. European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Arthr Rheumatol 2017;69:2271–2282. [CrossRef] [Google Scholar]
- Pinal-Fernandez I, Mammen AL. Dermatomyositis etiopathogenesis: a rebel soldier in the muscle. Curr Opin Rheumatol. 2018;30:623–629. [CrossRef] [PubMed] [Google Scholar]
- Marie I, Mouthon L. Therapy of polymyositis and dermatomyositis. Autoimmun Rev. 2011;11:6–13. [CrossRef] [PubMed] [Google Scholar]
- Marie I. Morbidity and mortality in adult polymyositis and dermatomyositis. Curr Rheumatol Rep. 2012;14:275–285. [CrossRef] [PubMed] [Google Scholar]
- Kim S, El-Hallak M, Dedeoglu F, Zurakowski D, Fuhlbrigge RC, Sundel RP. Complete and sustained remission of juvenile .dermatomyositis resulting from aggressive treatment. Arthritis Rheum. 2009;60:1825–1830. [CrossRef] [PubMed] [Google Scholar]
- Takada K, Katada Y, Ito S, Hayashi T, Kishi J, Itoh K, et al. Impact of adding tacrolimus to initial treatment of interstitial pneumonitis in polymyositis/dermatomyositis: a single-arm clinical trial. Rheumatology 2019. [Google Scholar]
- Mayu S, Isojima S, Miura Y, Nishimi S, Hatano M, Tokunaga T, et al. Polymyositis-dermatomyositis and interstitial lung disease in pregnant woman successfully treated with cyclosporine and tapered steroid therapy. Case Rep Rheumatol. 2019. [Google Scholar]
- Nawata T, Kubo M, Okuda S, Omoto M, Yujiri T, Kanda T, et al. Successful treatment with intravenous cyclophosphamide for anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis associated with myelodysplastic syndrome. Scand J Rheumatol. 2017;46:496–498. [CrossRef] [PubMed] [Google Scholar]
- Galimberti F, Kooistra L, Li Y, Chatterjee S, Fernandez AP. Intravenous immunoglobulin is an effective treatment for refractory cutaneous dermatomyositis. Clin Exp Dermatol. 2018;43:906–912. [CrossRef] [PubMed] [Google Scholar]
- Marie I, Menard J-F, Hatron PY, Hachulla E, Mouthon L, Tiev K, et al. Intravenous immunoglobulins for steroid-refractory esophageal involvement related to polymyositis and dermatomyositis: a series of 73 patients. Arthritis Care Res (Hoboken). 2010;62:1748–1755. [CrossRef] [PubMed] [Google Scholar]
- Chen KL, Zeidi M, Werth VP. Recent advances in pharmacological treatments of adult dermatomyositis. Curr Rheumatol Rep 2019;21:53. [CrossRef] [PubMed] [Google Scholar]
- Rodziewicz M, Kiely P. The successful use of subcutaneous abatacept in refractory anti- human transcriptional intermediary factor 1-gamma dermatomyositis skin and oesphagopharyngeal disease. Rheumatology 2018;57:1866–1867. [PubMed] [Google Scholar]
- Ioannou E. Thu0718-hpr overview of the most effective physiotherapy management methods in pediatric rheumatology. Ann Rheumatic Dis 2019;78:655–657. [Google Scholar]
- Goyal NA, Mozaffar T. Novel therapeutic options in treatment of idiopathic inflammatory myopathies. Curr Treatment Options Neurol. 2018;20:37. [CrossRef] [Google Scholar]
- Pearson DR, Werth VP. AB005. Unilateral vocal cord paresis in classic dermatomyositis. Ann Transl Med. 2019;7:AB005. [CrossRef] [Google Scholar]
- Traineau H, Aggarwal R, Monfort JB, Senet P, Oddis CV, Chizzolini C, et al. Treatment of calcinosis cutis in systemic sclerosis and dermatomyositis: a review of the literature. J Am Acad Dermatol. 2020;82:317–325. [CrossRef] [PubMed] [Google Scholar]
- Isak V, Jorizzo JL. Recent developments on treatment strategies and the prognosis of dermatomyositis: a review. J Dermatolog Treatm. 2018;29:450–459. [CrossRef] [Google Scholar]
- Cassius C, Le Buanec H, Bouaziz JD, Amode R. Biomarkers in adult dermatomyositis: tools to help the diagnosis and predict the clinical outcome. J Immunol Res. 2019;2019. [Google Scholar]
- Healy CM, Tobin AM, Kirby B, Flint SR. Oral lesions as an initial manifestation of dermatomyositis with occult malignancy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2006;101:184–187. [CrossRef] [PubMed] [Google Scholar]
- Gonçalves LM, Bezerra-Júnior JR, Gordón-Núñez MA, Libério SA, de Fátima V, Pereira A, et al. Oral manifestations as important symptoms for juvenile dermatomyositis early diagnosis: a case report. Pediatr Dent 2011;21:77–80. [CrossRef] [Google Scholar]
- Escobar A, Aitken-Saavedra JP. Xerostomia: an update of causes and treatments. In Salivary Glands-New Approaches in Diagnostics and Treatment 2018 Nov 5. IntechOpen. [Google Scholar]
- Adamczak MI, Martinsen OG, Smistad G, Hiorth M. Polymer coated mucoadhesive liposomes intended for the management of xerostomia. Int J Pharmaceut. 2017;527:72–78. [CrossRef] [Google Scholar]
- Gurkar H, Venkatesh OY, Somashekar JM, Gowda MH, Dwivedi M, Ningthoujam I. Prosthodontic management of xerostomic patient: a technical modification. Case Rep Dent 2016;2016. [Google Scholar]
- Geist SM, Tanaka TI. Oral lichen planus in a dermatomyositis patient that resolved after intravenous immunoglobulin therapy. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:e111–e114. [CrossRef] [PubMed] [Google Scholar]
- Ichimura Y, Matsushita T, Hamaguchi Y, Kaji K, Hasegawa M, Tanino Y, et al. Anti-NXP2 autoantibodies in adult patients with idiopathic inflammatory myopathies: possible association with malignancy. Ann Rheum Dis 2012;71:710–713. [CrossRef] [PubMed] [Google Scholar]
- Bassim CW, Gibson G, Ward T, Paphides BM, Denucci DJ. Modification of the risk of mortality from pneumonia with oral hygiene care. J Am Geriatr Soc 2008;56:1601–1607. [CrossRef] [PubMed] [Google Scholar]
- Rowen RJ. Remission of aggressive autoimmune disease (dermatomyositis) with removal of infective jaw pathology and ozone therapy: review and case report. Autoimmun Highlights 2018;9:1–7. [CrossRef] [Google Scholar]
- Sarifakioglu N, Terzioglu A, Ates L, Bingül F, Aslan G. Bilateral symmetric mandibular calcified nodules related to juvenile dermatomyositis. J Oral Maxillof Surg 2003;61:527. [CrossRef] [Google Scholar]
- Greer RO, Galbraith CT, Scheidt MJ, Aubry P, Glasgow MJ. Dermatomyositis with calcifications of the periodontal ligament: a rare oral finding. Dermatol Case Rep 2016;1:110. [CrossRef] [Google Scholar]
All Tables
All Figures
|
Fig 1 (A) Heliotrope rash of the eyelids. (B) The shawl sign over the upper back. (C) Gottron's papules over the extensor surfaces of the metacarpophalangeal and interphalangeal joints, with involvement of adjoining skin. (Reprinted from Chu LL, Rohekar G. Dermatomyositis. CMAJ. 2019; 191(12):E340. doi:10.1503/cmaj.180947, with permission). |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.